

Leukoencephalopathy with thalamus and brainstem involvement and high lactate (or LTBL)
Leukoencephalopathy with thalamus and brainstem involvement and high lactate (or LTBL) is an extremely rare genetic disorder in the family of leukodystrophies. Its prevalence is less than one in 1,000,000. It is one of the hypomyelinating leukodystrophies, those white matter pathologies characterised by a permanent deficit of myelin in the brain.
Because the syndrome is so rare, not much is known about the disease and very little information is available.
LTBL syndrome begins in early childhood. It is characterised by abnormalities in certain areas of the brain, including the thalamus and brain stem (the part of the brain that connects to the spinal cord), and by high levels of a substance called lactate in the brain and throughout the body. The syndrome usually leads to motor problems and problems controlling muscle function.
The gene whose mutation is responsible for LTBL syndrome is the EARS2 gene which is located on chromosome 16 (at 16p12.2). This gene codes for a protein, glutamyl-tRNA synthetase , which is involved in the manufacture of proteins in mitochondria.
Protein synthesis: an industry!
In cells, an entire industry is responsible for protein synthesis. These molecules, diverse and numerous in the cell, perform a multitude of roles. Proteins are all synthesised according to the same principle, in the same plants, but each one is made following the instructions specific to that protein which are contained in the genome in the form of a DNA sequence.
The genome is hidden in the cell nucleus, a veritable library storing the recipes for making all proteins. Based on these instructions, a copy of the recipe will be reproduced in RNA form, to be sent to the unit in charge of production. Production units then synthesise the proteins by assembling the building blocks following the instructions reproduced in the RNA.
PRODUCTION SITE
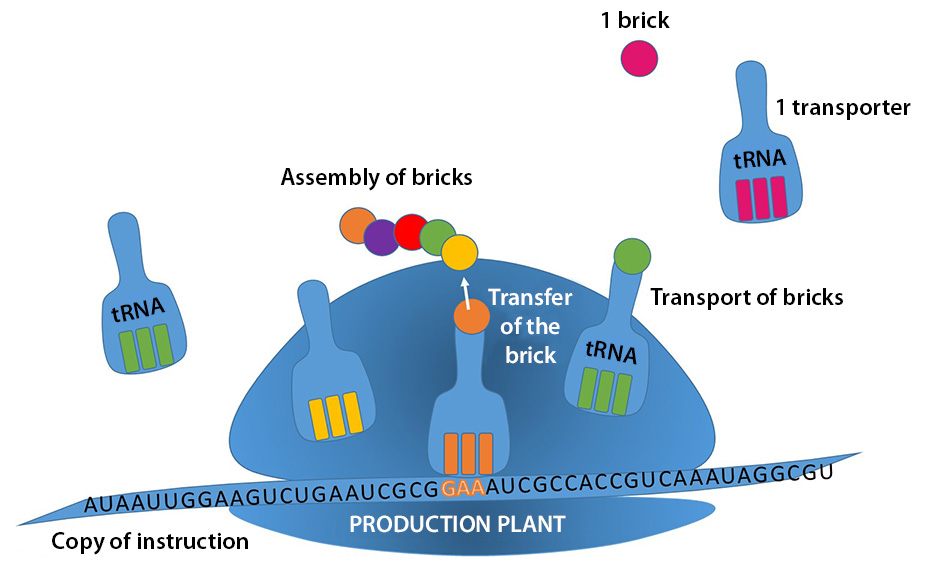
The tRNA gets its name from its function of transferring the blocks (amino acids) necessary for building proteins. It is this conveyor that brings the material into the production area, then allows it to be attached to the bricks already assembled.
The tRNA synthetase is responsible for fixing the right brick to the tRNA. The preparation of this specialised worker and therefore the whole production chain in general depends on it.
Symptoms of the disease
It was in 2012 that a team of researchers was able to put a name to the gene that is mutated in LTBL syndrome. To do this, the researchers analysed the DNA of 12 patients with the same characteristics through next-generation exome sequencing. The disease is so rare that to carry out this work, the young patients came from all over the world: one Italian, two Belgians, two from England, one American, one Israeli, one Swiss, one Portuguese, one German, and two Brazilians.
Forms of the disease
Patients with LTBL syndrome all have symptom onset in infancy and rapid disease progression with severe abnormalities visible in MRI results and increased lactate levels. The disease comes in two forms: a mild form and a severe form.
In its mild form, from 6 months of age, the disease leads to a loss of mental and movement abilities (psychomotor regression). Muscle stiffness (spasticity) and extreme irritability are common, and some people develop seizures. Patients then make a partial recovery. Developmental milestones may be delayed, but children are able to acquire new abilities in the years that follow. MRI results shows striking improvements and lactate levels are dropping.
For patients with the severe form of the disease, symptoms begin soon after birth. These infants generally have delayed development of mental and movement abilities (psychomotor delay), weak muscle tone (hypotonia), involuntary muscle tensing (dystonia), muscle spasticity and seizures. Some have extremely high levels of lactate (lactic acidosis), which can cause serious respiratory and heart problems. Liver failure occurs in some severely affected infants. Subsequently, clinical stagnation, brain atrophy visible on MRI and a persistent increase in lactate levels are noted.
Diagnosis of the disease
A diagnosis can now be suggested by the patient’s clinical picture, and the characteristics of an MRI image. A rise in lactate levels is also measurable and indicative. To confirm the diagnosis, sequencing of the EARS2 gene will show a mutation in each of the two copies of the gene (autosomal recessive disease: parents each carry one copy of the mutated gene, but they usually do not show the signs and symptoms of the disease).
The simplified mechanism leading to the disease
A mitochondrion is a separate structure in the cell, responsible for ensuring the cell’s respiration, and thus producing energy for the cell. Mitochondria use their own protein synthesis system, their own plant, to ensure their own operations. But the principle remains the same (see diagram: Making proteins: an industry!). The tRNA synthetase from the EARS2gene allows glutamate (the brick) to be fixed to its transfer RNA which works in the mitochondria.
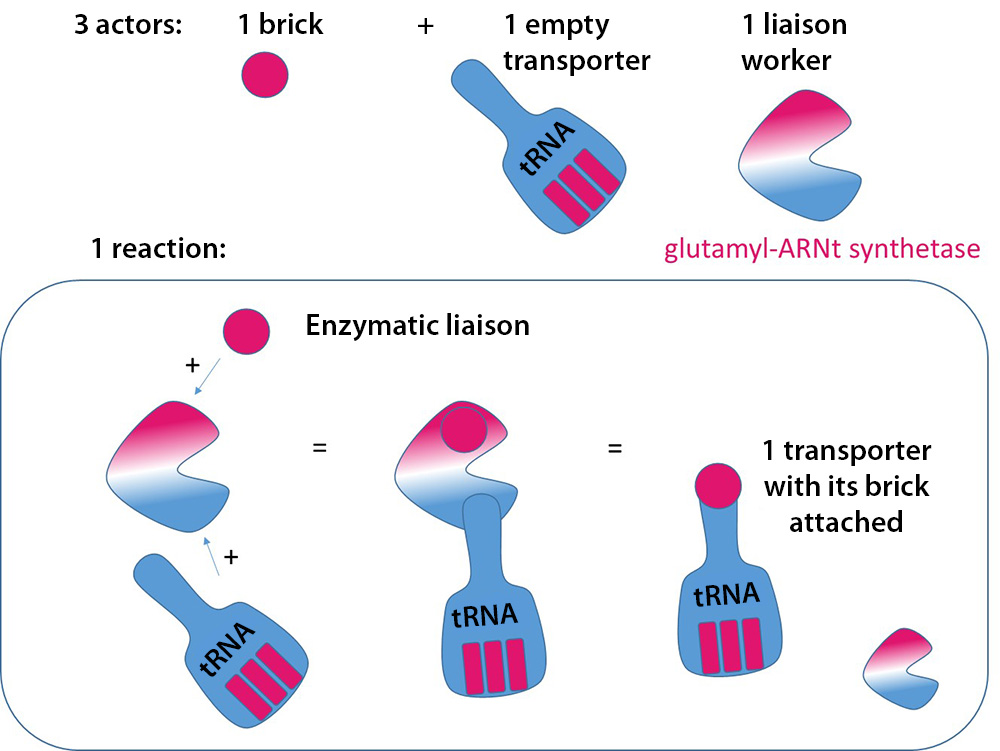
Enzymatic binding of amino acids to tRNAs: glutamyl-tRNA synthetase , derived from the EARS2 gene, is the enzyme (the worker) needed to assemble the amino acid glutamate (the brick) on the tRNA (the transporter).
In LTBL syndrome, researchers have been able to identify the gene that is mutated, and believe that the amount of glutamyl-tRNA synthetase is reduced in patients. The reduced amount of the enzyme probably prevents the normal assembly of new proteins in the mitochondria. It is suggested that this disrupts the mitochondria’s energy production. However, it is not clear today how mutations in the EARS2 gene lead to the clinical features of LTBL syndrome.
Treatments
Day-to-day management of the disease
Treatment is symptomatic and is ideally conducted in a multidisciplinary setting by specialists experienced in treating people with leukodystrophies. Medication is available to manage muscle tone. Intensive physiotherapy can be used to improve mobility and function. The treatment of ataxia, seizures and cognitive problems follows the usual standards, depending on the needs of the individual.
Monitoring should be maintained to assess the child’s growth and nutritional status. Serial physical and/or radiographic examinations of the hips and spine can be used to monitor orthopaedic complications. Past history is made up of the signs and symptoms of seizures.
When a disease is so rare, how can you hope to cope with it and put a name to what affects your child? Mrs Karine Garest, Marion’s mother, tells us how she was finally able to do it by listening to the description of the disease given by Prof. van der Knaap during an ELA family/researcher conference.
“In 2012, during the symposium organised by ELA, I attended the workshop on undetermined leukodystrophies where Prof. van der Knaap presented a new form of leukodystrophy, LTBL. In the description she described a lactate attack in the brain between the ages of 0 and 2 that more or less destroyed the myelin, and that there was a high level of lactate in the blood. Then I remembered a blood test that Marion had taken when she was around 9 months old and she had a high lactate level. This started to send me back to our story. Then she shared the children’s development saying that after this attack there was no more regression and that on the contrary the children were progressing and this was the case for Marion since she sat up at 2 years old, walked at 5 years old etc… so I recognised Marion’s condition in this description and we were able to talk at the end of the workshop and communicate through email afterwards until the blood test which confirmed that Marion was suffering from this form of leukodystrophy……
At the moment, she is the only one who has been diagnosed in France, which, as Prof. Wolf at the symposium, is not possible, there are certainly other people with this form of leukodystrophy, so I hope that my testimony will allow other families to put a name to their child’s disease because if I had not been at the symposium that year, Marion’s condition would still be part of the undetermined leukodystrophies …. ”
