

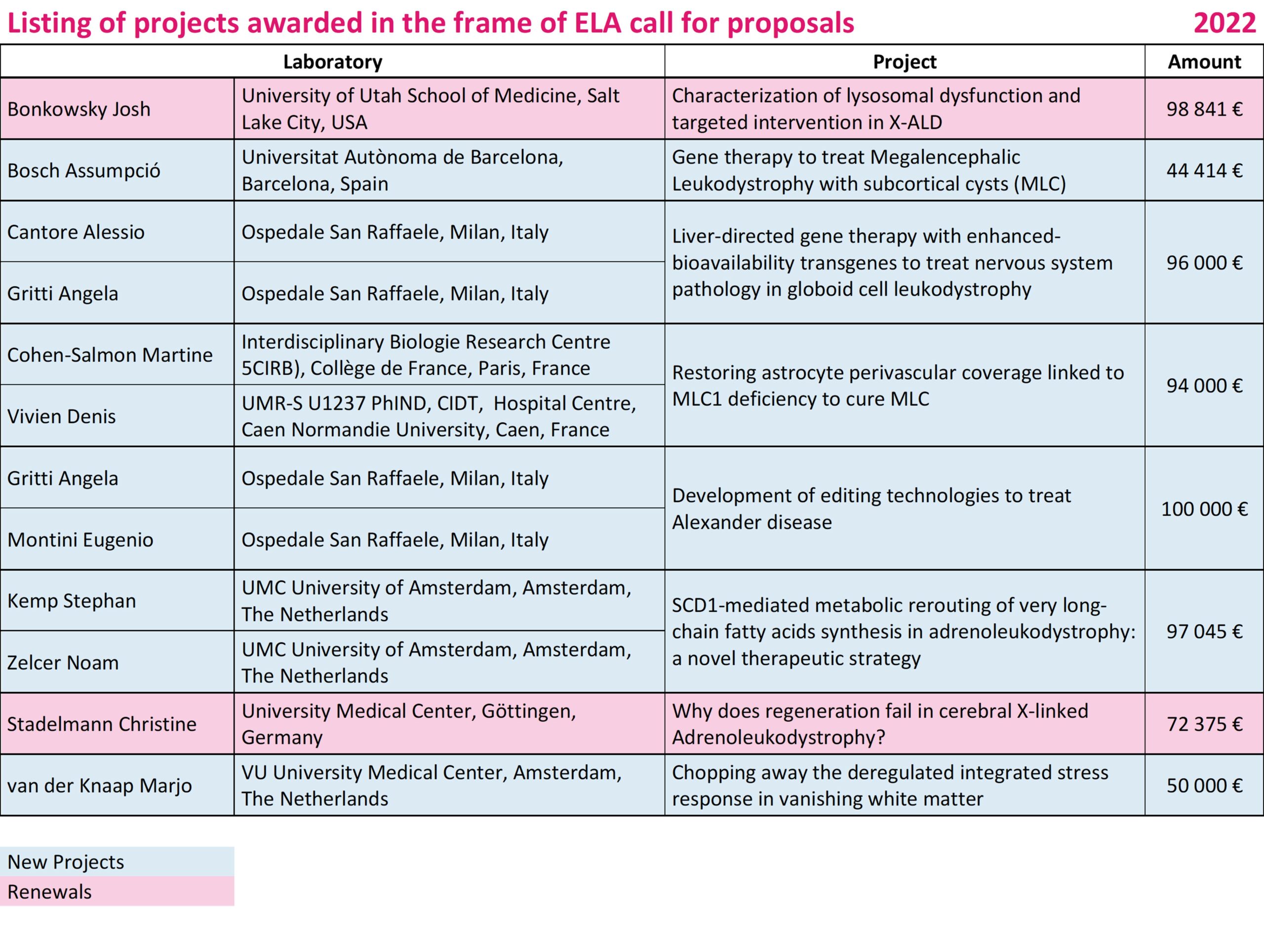
Bosch Assumpció • Spain
2022-004I2
Gene therapy to treat Megalencephalic Leukodystrophy with subcortical cysts (MLC)
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a rare genetic disorder characterized by an abnormal head, loss of motor functions, epilepsy and mild mental decline. The disease is caused by mutations in two genes named MLC1 and GLIALCAM. There are no therapies for MLC patients, only palliative treatment. We recently demonstrated the efficiency of gene therapy in correcting MLC-typical myelin vacuolation. Now we aim to demonstrate that gene therapy is also able to correct the motor impairment present in the MLC knock-out mouse models. Thus, we propose to administer the therapeutic AAV vectors to the affected cells in the brain of these mice, and to look for longitudinal, non-invasive tests to assess for in vivo indicators of disease correction in the animal models that could finally be translated to clinical trials for MLC patients.
Cantore Alessio / Gritti Angela • Italy
2022-006C2
Liver-directed gene therapy with enhanced-bioavailability transgenes to treat nervous system pathology in globoid cell leukodystrophy
Globoid cell leukodystrophy (GLD) is a neurodegenerative inherited genetic disease, due to defects in an enzyme that normally degrades sphingolipids, a class of biological molecules present in the myelin sheath, necessary for nervous tissue function. The name of this enzyme is beta-galactosylceramidase (GALC). Impairment of GALC activity in GLD results in progressive demyelination and nerve tissue degeneration. Most individuals with infantile forms die before the age of two. Currently, there is no cure for GLD.
Several gene replacement therapy strategies have been attempted in pre-clinical models with varying degree of success, however, still facing the challenge of providing widespread enzymatic reconstitution and full correction of pathology in affected tissues, particularly the central and peripheral nervous systems (CNS and PNS). Previous evidence suggests effective transport across the blood brain barrier of several therapeutic proteins when fused with parts of other proteins, known to be able to reach CNS and PNS. Thus, GALC will be endowed with these additional portions, which may be sufficient to ensure its transport across the blood brain barrier.
In addition, we will equip GALC with small portions of highly secreted proteins to improve GALC secretion by cells. The long-term objective of the proposed research is thus to develop a safe and effective liver gene therapy for GLD. In vivo gene therapy to the liver indeed offers the attractive prospects of a minimally invasive, one-time potentially curative treatment for GLD, by providing sustained pervasive high amounts of a functional GALC enzyme to the CNS and PNS through the bloodstream.
Here we propose a project to obtain an early proof-of-principle in a mouse model of the disease. The CNS and PNS are naturally protected by a blood brain barrier that ensures selective passage of necessary substances, while blocking potentially dangerous molecules. Lentiviral vectors (LV) are attractive gene delivery vehicles for liver gene therapy thanks to their ability to insert their DNA into the host cell DNA. For this reason, LV are maintained following liver cell proliferation in liver growth and turnover, including potentially in newborns, that would be the target population in GLD. We have developed LV that allow stable expression of therapeutic proteins in the liver of mice and dogs, following systemic administration. More recently, we have generated engineered LV with increased resistance to capture by cells of the immune system, a process called phagocytosis. Thanks to this feature, these LV are more efficient at reaching the target cells in the liver.
Here we propose to generate LV expressing the above-described optimized GALC from the liver, administer them intravenously to newborn mice affected by GLD and assess survival and correction of the hallmarks of the GLD disease. We also propose to engineer LV to further escape phagocytosis by exposing on their surface additional inhibitors of phagocytosis. If successful, the work proposed here will allow the generation of novel LV with increased protection from phagocytosis and efficiency of gene transfer into liver cells that will facilitate development and manufacturing to quality and scale required for use in humans and alleviate concerns of possible LV particle dose dependent acute toxicity. These LV, transporting the engineered GALC enzymes, may become a feasible, sustainable, safe and effective liver gene therapy for GLD and potentially for other leukodystrophies.
Cohen-Salmon Martine / Vivien Denis • France
2022-007C4
Restoring astrocyte perivascular coverage linked to MLC1 deficiency to cure MLC
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a rare disease mainly related to mutations in the MLC1 gene. Patients suffer from macrocephaly and motor and cognitive defects associated with progressive myelin degeneration. We have recently shown that MLC originates from early defects in the gliovascular unit, a specialized interface in the brain between blood vessels and astrocytic glial cells, including a defect in vessel contractility that impairs blood flow in the brain. We believe that these alterations are at the origin of MLC. Our project is now to develop an approach to restore the functions of the altered gliovascular unit in a mouse model of MLC and to test whether this strategy can reverse leukodystrophy, which would pave the way for therapeutic intervention in patients.
Gritti Angela / Montini Eugenio • Italy
2022-009C2
Development of editing technologies to treat Alexander disease
Alexander disease (AxD) is a rare autosomal dominant disorder caused by point mutations in the gene encoding for the glial fibrillary acidic protein (GFAP), the major intermediate filament protein in astrocytes. Accumulation of GFAP protein in Rosenthal fibers leads to astrocytic dysfunctions and altered development and homeostasis of affected brain tissues. No cures are currently available for AxD patients.
In the proposed pilot project, we aim to develop gene-editing approaches to downregulate the expression of the mutated GFAP protein or correct mutations in the Gfap gene in an animal AxD model. This project will collect solid proof-of-concept data for the future progress of a novel and definitive gene therapy strategy for the treatment of AxD patients. In addition, we expect to outline novel editing platforms that could be applied prospectively for disease modeling studies and treatments of other leukodystrophies characterized by astrocyte degeneration or dysfunctional/maladaptive astrogliosis
Kemp Stephan / Zelcer Noam • The Netherlands
2022-011C2
SCD1-mediated metabolic rerouting of very long-chain fatty acids synthesis in adrenoleukodystrophy: a novel therapeutic strategy
X-linked adrenoleukodystrophy (ALD) is the most common leukodystrophy. All ALD patients have a mutation in the ABCD1 gene that leads to a build-up of saturated very long-chain fatty acid (VLCFA) in tissues, including adrenal glands, spinal cord and brain. ALD is characterized by a striking and unpredictable clinical spectrum, even within families. In childhood, around 50% of affected boys develop adrenal disease before the age of 10 and 30-35% of affected boys develop a fatal inflammatory brain disease (cerebral ALD). If ALD is diagnosed in an early stage, cerebral ALD can be halted or reversed by a bone-marrow transplant. In adulthood, virtually all males and >80% of women develop a chronically slow progressive spinal cord disease (myeloneuropathy) for which no disease modifying therapy is available. Unfortunately, transplanted boys can still develop spinal cord disease in adulthood, because the transplant is only effective at halting the inflammatory component of the disease without addressing the underlying biochemical defect. This therapeutic gap highlights the need to develop effective treatments aimed at the normalization of VLCFA levels in the brain and spinal cord.
Using skin cells from ALD patients we have demonstrated that saturated VLCFA induce cellular stress, with prolonged exposure resulting in cell death. Remarkably, this is not observed with mono-unsaturated VLCFA (fatty acids with a double bond in the fatty acid chain). The enzyme stearoyl-CoA desaturase-1 (SCD1) coverts saturated fatty acids into mono-unsaturated fatty acids. In a previous ELA supported project we identified and characterized the drug TO901317 (an LXR agonist) as a small-molecule that activates SCD1 activity. Treatment of ALD cells with TO901317 completely corrects VLCFA levels and treatment of the ALD mouse with TO901317 added to the food resulted in a reduction in VLCFA levels in adrenals, spinal cord and brain.
Unfortunately, TO901317 (and other LXR agonists) as potential treatment for ALD have notable limitations due to off-target effects that result in serious side effects. This is largely due to LXR agonists not being specific for SCD1. We therefore hypothesize that specifically increasing SCD1 activity will circumvent the detrimental effect of LXR activation and offer a therapeutic strategy to counteract VLCFA-induced lipotoxicity in ALD.
Strategies to specifically increase SCD1, either pharmacologically or genetically have not been reported to date. Therefore, the overall goal of the proposal is to identify genetic and pharmacological regulators of SCD1 abundance and/or activity. Specifically, in this project we aim to:
- Generate cell lines in which the SCD1 enzyme is tagged with a fluorescent protein to allow monitoring of SCD1 in
- live cells at a single cell resolution.
- Map enzymes that control SCD1 abundance using a genome-wide CRISPR/Cas9 genetic screen.
- Identify small molecules that increase SCD1 abundance and activity. Addressing these aims will increase our basic understanding of VLCFA metabolism in ALD.
Moreover, results from these experiments have the potential to inform on novel therapeutic strategies to treat ALD patients.
van der Knaap Marjo • The Netherlands
2022-018I2
Chopping away the deregulated integrated stress response in vanishing white matter
Leukodystrophies are a major source of handicap at all ages, but children are affected most. We have studied leukodystrophies since 1987. Initially, our research focused on describing new leukodystrophies and finding the underlying gene defects. Much of our subsequent research efforts concerned the new leukodystrophy vanishing white matter (VWM), also called childhood ataxia with CNS hypomyelination (CACH). The disease occurs at all ages, but mainly starts in young children (2-6 years). Children with VWM experience progressive neurological handicap that prevent fever and head trauma, as these events trigger a fast worsening of the disease.
Several years ago we found that the gene defect for VWM lies in an enzyme complex that is crucial for protein synthesis. Since then we have studied how the disease works (disease mechanisms), most of all to find openings for potential treatment. We found that cells in the white matter of the brain do not develop into mature cells that can execute their normal function of myelination and white matter repair properly. The problem with maturing into functional cells can well explain the severe white matter damage that we observe in patients. Our recent studies have demonstrated that a basic stress pathway is abnormally activated in VWM white matter cells. We have evidence that abnormal activation of this stress pathway causes the disease. In the proposed study we will target and inhibit an important and toxic component of this stress pathway with antisense oligonucleotides and test the effects in a representative disease model. The proposed work has the potential to open up new treatment options relatively fast.
Our vision on treatment of VWM is that effective treatment of this complex disease is most likely not achieved with any single therapeutic modality. We think that the treatment should target the disease at multiple levels, including reduction of stress pathways, reduction of the toxicity of the diseased white matter, provision of healthy white matter cells and possibly gene therapy.
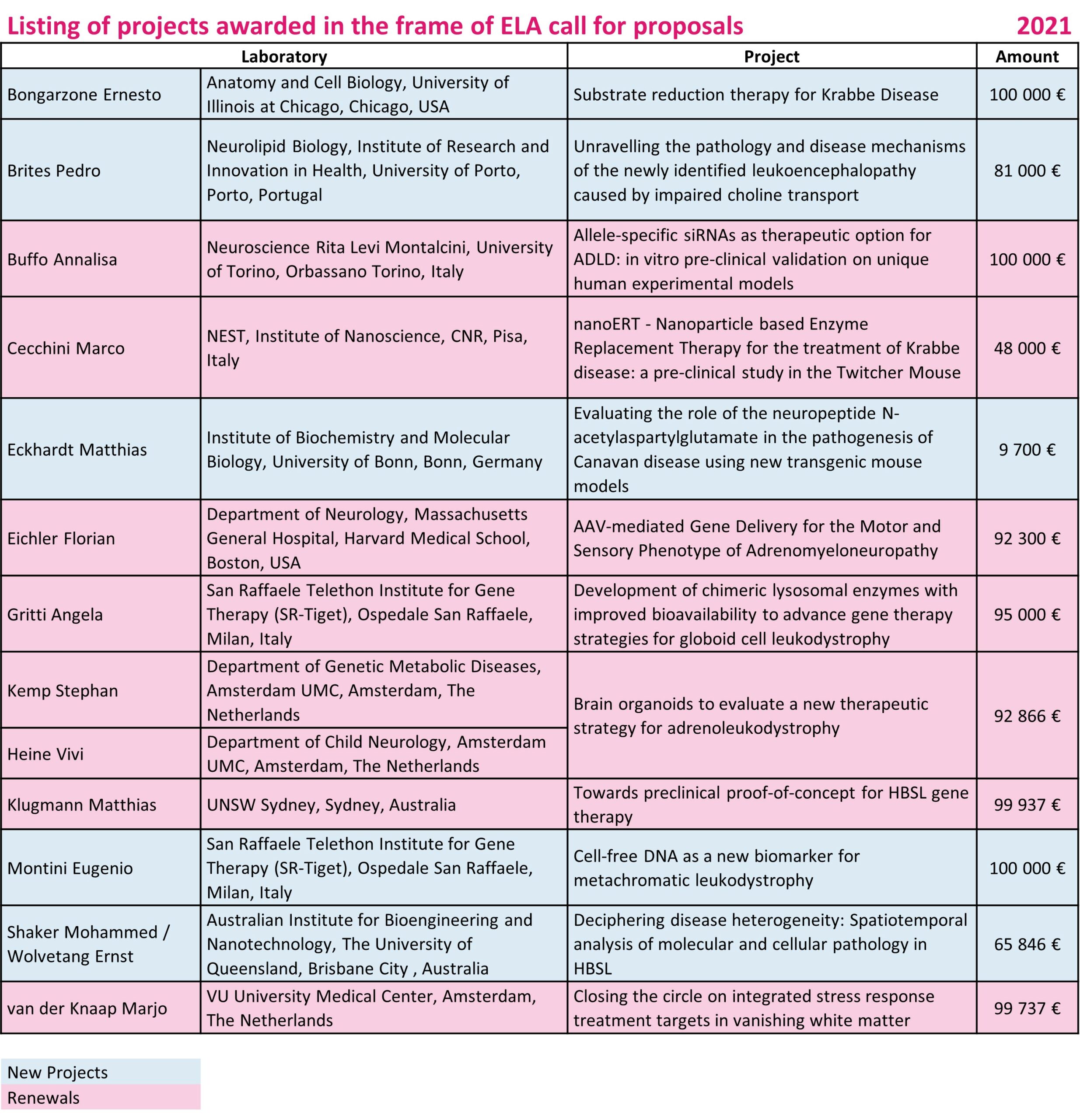
Bongarzone Ernesto • USA
2021-002I2
Anatomy and Cell Biology University of Illinois at Chicago
This project will test a new small molecule that is designed to reduce the activity of the enzyme acid ceramidase, which is the enzyme that synthesizes psychosine. Psychosine is a lipid that accumulates to high levels in the brain of Krabbe disease, a genetic disorder caused by deficiency of another enzyme called GALC, which controls the degradation of psychosine. In the absence of GALC activity, psychosine continues to be produced but not degraded. High levels of psychosine in Krabbe disease cause neuropathology and motor and cognitive declines in infants, juvenile and adult patients. There is no cure for Krabbe disease. Here we will treat animal models of infantile and adult onset Krabbe disease with the small compound singly and in combination with a gene therapy approach to restore the activity of the enzyme GALC, which is deficient in Krabbe disease. We hypothesize that reducing the synthesis of psychosine will improve the metabolism of psychosine, reduce disease burden and neuropathology.
Brites Pedro • Portugal
2021-004I3
Unravelling the pathology and disease mechanisms of the newly identified leukoencephalopathy caused by impaired choline transport
For its correct development and function, nervous tissue synthesizes various key components but is also reliant on essential nutrients. Choline is one of such nutrients that is necessary for the synthesis of phospholipids and the generation of the neurotransmitter acetylcholine. A novel disorder was recently identified and shown to be caused by mutations on SLC44A1, the gene that encodes the choline transporter-like protein 1. With an early childhood-onset presentation, this novel leukoencephalopathy is characterized by severe white matter involvement, optic nerve atrophy, ataxia, dysarthria, tremors, and patients have delayed motor and speech development. In order to have a valid animal model for the disorder, we generated the first Slc44a1 mutant mice to characterize the underlying pathology and disease mechanisms caused by a defect in choline transport. Using the Slc44a1 mutants, we will determine how choline dysregulation impairs oligodendrocyte differentiation, myelination, and neuron function. Our aims are to: 1- Determine the neuropathology caused by impaired choline transport; 2- Establish the proteomic and metabolic changes caused by choline deficiency; and 3- Evaluate the therapeutic potential of choline supplementation. Combined, this project addresses several unmet scientific and medical needs that are set to have a significant beneficial impact on scientific and societal communities.
Eckhardt Matthias • Germany
2021-009I4
Evaluating the role of the neuropeptide N-acetylaspartylglutamate in the pathogenesis of Canavan disease using new transgenic mouse models
Deficiency in the enzyme aspartoacylase leads to a severe leukodystrophy, called Canavan disease, which is characterized by the accumulation of the brain metabolite N-acetylaspartate (NAA). NAA is synthesized and released mainly by neurons. Many neurons use NAA also to synthesize the small neuropeptide N-acetylaspartylglutamate (NAAG). Extracellular NAAG secreted by neurons can be degraded to NAA and glutamate by an enzyme expressed by astrocytes. Thus, degradation of NAAG is another source of NAA. The current project will further investigate the role of NAAG in the NAA accumulation observed in Canavan disease. This will be done using different transgenic mouse models, including a mouse model of Canavan disease, that differ in their capability to synthesize NAAG. In the project, histological and biochemical methods will be used to evaluate the influence of the genetic changes in these mouse models on the accumulation of NAA and the development of brain spongiform degeneration and myelin loss. The aim of this project is to examine whether reduced NAAG synthesis leads to reduced NAA accumulation and reduced pathological changes in the Canavan disease mouse model. If this would be the case, inhibition of NAAG synthesizing enzymes would potentially be an alternative or additional option for a substrate reduction therapy of Canavan disease.
Montini Eugenio • Italy
2021-019I1
Cell-free DNA as a new biomarker for metachromatic leukodystrophy
Metachromatic Leukodystrophy (MLD) is a genetic disorder caused by mutations in the ARSA gene. The absence of this enzyme leads to accumulation of sulfatides in neural and glial cells leading to progressive neurodegeneration and eventually death. Our firstin-human clinical trial for the treatment of MLD based on hematopoietic stem cell (HSC) gene therapy (GT) provided evidences on the feasibility, safety and efficacy of the procedure prompting its authorization with the name of Libmeldy. With this therapy, patients’ HSCs are genetically corrected and reinfused to reconstitute the entire hematopoietic system acting as “Trojan horses” for the delivery of the ARSA enzyme to the affected tissues. This treatment provide benefit when provided to pre- or early symptomatic MLD patients. Unfortunately, it is difficult to predict if a candidate MLD patient with genetic mutations of the ARSA gene will ever develop symptoms of the disease. Therefore, the clinical decision making for treatment is based on the case-by-case evaluation of patient’s clinical history and several clinical biochemical and instrumental assays. Moreover, treated patients must be monitored for the safety and efficacy of the therapy which include additional molecular analyses addressing the clonal composition and behavior of genetically modified blood cells after transplantation. Although these analyses are extremely useful to define the efficacy and safety of GT, the identification of additional biomarkers more predictive of disease burden and useful as treatment response indicators is strongly needed.
We recently developed LiBIS-seq (liquid-integration-site-sequencing), a PCR technique optimized to exploit blood plasma cell free DNA (cfDNA) for the study of the clonal composition of genetically modified cells in circulation and in solid tissues. cfDNA is constituted by short DNA fragments released in body fluids by dying cells residing in different tissues of the body and for this reason its analysis provided more reliable safety predictions of the GT approaches than the methods based only on the analysis of circulating cells. Moreover, cfDNA concentrations in plasma of MLD GT patients were above pathological levels before the treatment and decrease progressively after transplantation in patients with a positive therapeutic outcome. Hence, the level of cfDNA in MLD patients may reflect the number of dying cells because of the accumulation of toxic lysosomal compounds.
For these reasons, we aim to validate blood plasma cfDNA as new and non-invasive outcome measure of disease severity and as surrogate biomarker of the efficacy of GT treatment for MLD. Specifically, we will use state of the art technologies to investigate the epigenome an integrome on cfDNA and cells collected overtime from early juvenile and late infantile MLD patients treated by GT in our institution. These analyses will define the efficacy and safety of the treatment at the clonal level, determine which cell types are contributing to cfDNA production in MLD GT patients and in natural history patients unravelling tissue-specific toxicities over the course of the disease. If successful, the identification of a reliable disease biomarker will allow for a more accurate diagnosis evaluation and a more personalized therapeutic intervention for MLD.
Shaker Mohammed (PI: Ernst Wolvetang) • Australia
2021-024F2 (Fellow)
Deciphering disease heterogeneity: Spatiotemporal analysis of molecular and cellular pathology in HBSL
In Hypomyelination with Brain stem and Spinal cord involvement and Leg spasticity (HBSL), the rate and degree of spread of symptoms throughout the body are linked to survival outcome. However, the cause for the pathology in HBSL is unknown. Using mini 3D brain oligodendrocytes generated from HBSL patient skin cells, we will study how interactions between oligodendrocytes and their support cells (neurons, astrocytes, and microglia) might lead to the development of disease over time. We will apply the cutting-edge technology of spatial transcriptomics to generate “cell-to-cell network maps” to inform whether we can manipulate cell networks to prevent demyelination. Finally, this research project aims to test nutraceutical supplementation as a therapeutic potential to ameliorate and or reverse established HBSL pathology.
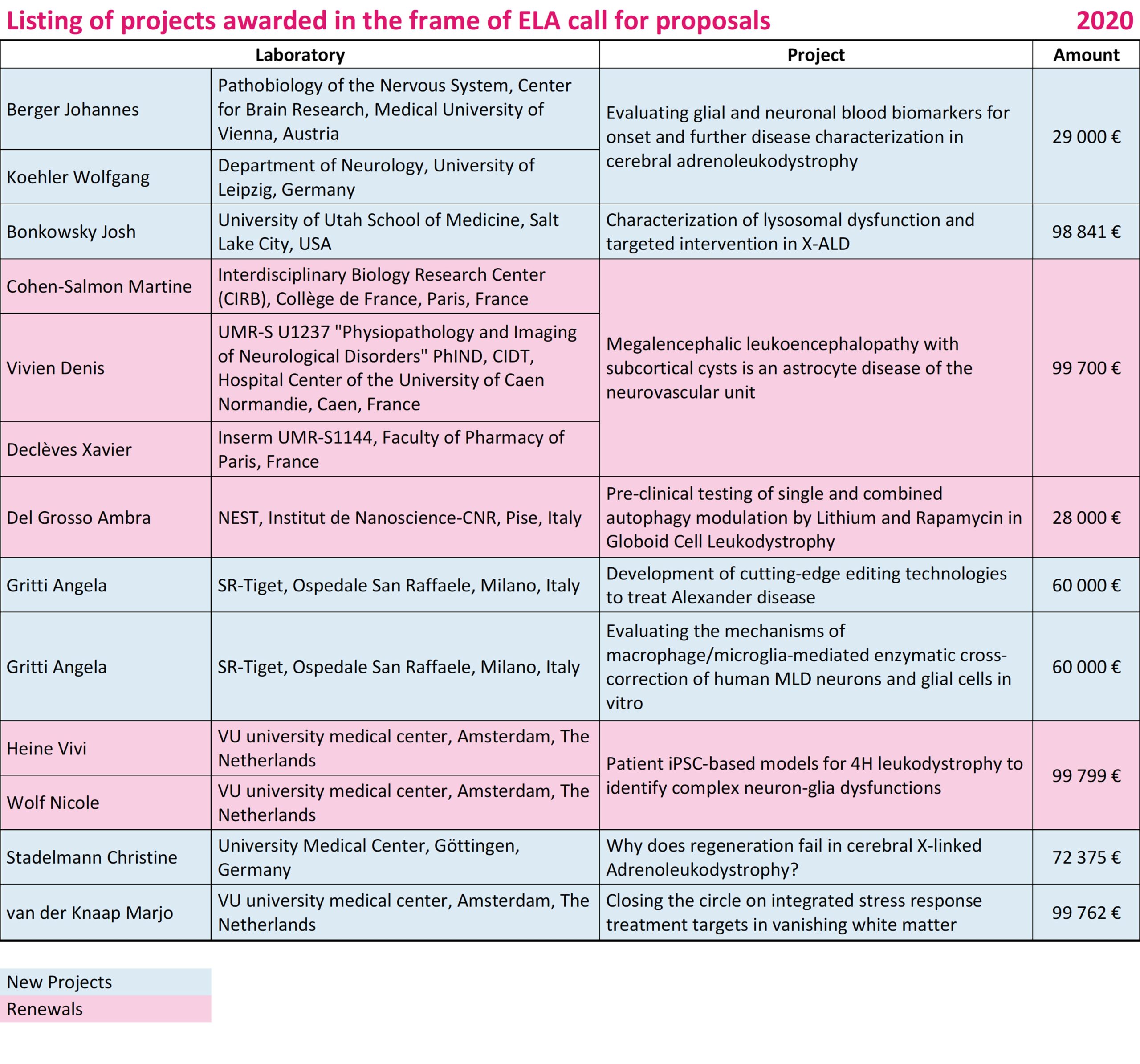
Berger Johannes / Koehler Wolfgang • Austria / Germany
2020-003C1
Evaluating glial and neuronal GFAP, MCP-1 and NfL as blood biomarkers for onset and disease severity in cerebral adrenoleukodystrophy
With a combined incidence of 1:14,700, X-ALD is the most common monogenetically inherited leukodystrophy. The disease is caused by mutations of the peroxisomal very long-chain fatty acid transporter ABCD1 that normally imports very long-chain fatty acids into the peroxisome for degradation. Accordingly, loss of ABCD1 function results in accumulation of very long-chain fatty acids in the plasma and body fluids of affected patients. X-ALD shows a striking phenotypic heterogeneity with inflammatory cerebral X-ALD (CALD) being the most severe form. In order to be treatable by bone marrow transplantation or gene therapy, CALD has to be recognized in its earliest stages.
The ultimate goal of this research project is to identify an easily accessible blood biomarker indicative for onset and progression of CALD. If successful, the identified blood biomarker could provide valuable information for decisions on clinical interventions and could also be used as treatment efficacy marker in clinical trials targeting CALD.
Bonkowsky Josh • USA
2020-004I3
Characterization of lysosomal dysfunction and targeted intervention in X-ALD
X-linked adrenoleukodystrophy (ALD) is a severe disease, ranging from the fatal cerebral inflammatory demyelinating form affecting boys (cALD) to the peripheral dying back axonopathy that affects adults (AMN). There are no effective therapies for AMN, and the cause of cALD is poorly understood.
The goal is to develop, validate, and use for treatment discovery, a new model of ALD. To accomplish this, the group will use the small genetic vertebrate model zebrafish, which has low costs and the ability to perform high-throughput screens that are unfeasible in other vertebrate systems. Zebrafish have the same genes as humans, including ABCD1, the gene responsible for ALD. The data shows that mutations in zebrafish ABCD1 cause ALD in zebrafish. The team is using the ALD zebrafish model to validate a new potential treatment for ALD, to understand this drug’s mechanism, and to understand the cause of cALD.
Gritti Angela • Italy
2020-010I2
Development of cutting-edge editing technologies to treat Alexander disease
Alexander disease (AxD) is a rare autosomal dominant disorder caused by point mutations in the gene encoding for the glial fibrillary acidic protein (GFAP), the major intermediate filament protein in astrocytes. Accumulation of GFAP protein in Rosenthal fibers leads to astrocytic dysfunctions and altered development and homeostasis of affected brain tissues.
No cures are currently available for AxD patients. In the proposed pilot project, the team aim to develop a gene editing approach to specifically downregulate the expression of the mutated GFAP protein in affected AxD astrocytes. This project will collect solid proof-of-concept data for the future progress of a novel and definitive gene therapy strategy for the treatment of AxD patients. In addition, they expect to outline novel editing platforms that could be applied prospectively for disease modelling studies and therapeutic treatments of other leukodystrophies characterized by astrocyte degeneration or dysfunctional/maladaptive astrogliosis.
Gritti Angela • Italy
2020-011I2
Evaluating the mechanisms of macrophage/microglia-mediated enzymatic cross-correction of human MLD neurons and glial cells in vitro
Hematopoietic stem cell gene therapy (HSC GT) is an experimental treatment based on the transplantation of autologous hematopoietic precursor cells genetically modified to express high levels of ARSA enzyme. This treatment benefits late infantile and early juvenile MLD children if treated in the pre-symptomatic/early symptomatic stage of the disease but is less effective in MLD children treated in the progressive phase of disease, who experience severe neurological deterioration. In order to recognize the reasons behind this different clinical outcome we need a better understanding of the therapeutic mechanisms of correction of MLD neural cells by the progeny of transplanted hematopoietic cells that engraft in the brain.
In this project the investigators aim to fill this gap of knowledge taking advantage of unique and clinically relevant in vitro human disease models, i.e. neural and blood cells derived from healthy donors, untreated and gene therapy treated MLD patients. We expect to clarify how and to which extent the ARSA enzyme is transported from metabolically competent donor blood cells to MLD neurons and glia. Also, they will provide hints on the contribution of complementary mechanisms of correction that could be exploited to enhance the benefit of HSC GT and broaden its accessibility to larger cohorts of MLD patients who presently not meet the inclusion criteria of these experimental treatments.
Stadelmann Christine • Germany
2020-016I4
Why does regeneration fail in cerebral X-linked adrenoleukodystrophy?
X-linked adrenoleukodystrophy is a genetic disease affecting mostly young male patients. In its severest form, patients develop a fulminant inflammatory destruction of central nervous system white matter, areas where all descending nerve fiber tracts are contained. Untreated, patients suffering from this disease mostly die within months to few years. The only effective therapy for halting the progression is an early transplantation of hematopoietic stem cells. Though progression often can be stopped this way, any disability that patients had accumulated before treatment recovers very poorly. This is in harsh contrast to another inflammatory disease affecting the white matter, multiple sclerosis. Here, patients often recover very well after an inflammatory and demyelinating bout. The reasons for this difference are not yet understood.
In this project, the team strive to investigate the reasons underlying the apparent lack of regeneration in X-ALD. They will use autopsy tissue from human patients who died of X-ALD as a starting point and let their research be guided by a careful re-examination of pathologic changes in this tissue. In addition, they will use novel proteome and transcriptome techniques that allow a very detailed investigation of molecular alterations in the tissue. In this way the researchers strive to identify novel molecular and cellular pathways of the disease that could be manipulated in order to improve clinical recovery in X-ALD. Furthermore, in their novel detailed analysis of the human histopathology they hope to provide a valuable tool also for other researchers working in the field.
van der Knaap Marjo • The Netherlands
2020-017I2
Closing the circle on integrated stress response treatment targets in vanishing white matter
Leukodystrophies are a major source of handicap at all ages, but children are affected most. The team studies leukodystrophies since 1987. Initially their research was focused on describing new leukodystrophies and finding the underlying gene defects. Much of their research efforts have focused on the new leukodystrophy vanishing white matter (VWM), also called childhood ataxia with CNS hypomyelination (CACH). The disease may occur at all ages, but mainly affects young children (2-6 years). Children with VWM experience progressive neurological handicap and die early, usually a few years after diagnosis. There is no cure for VWM, but patients benefit from treatments that prevent fever and head trauma, as these events trigger a fast worsening of the disease.
Several years ago, the team founds that the gene defect for VWM lies in an enzyme complex that is crucial for protein synthesis. Since then, they study how the disease works (disease mechanisms), most of all to find openings for treatment. The group founds that cells in the white matter of the brain do not develop into mature cells that can execute their normal function of myelination and white matter repair properly. The problem with maturing in functional cells can well explain the severe white matter damage that is observed in patients. Their recent studies have demonstrated that a basic stress pathway is abnormally activated in VWM white matter cells. They have evidence that abnormal activation of this stress pathway may contribute to disease.
In the proposed study they will test the effects of three FDA-approved inhibitors of this stress pathway in representative disease models. The proposed work has the potential to open up new treatment options fast. Their vision on treatment of VWM is that effective cure of this complex disease is not achieved with any single therapeutic modality. The team believes that the treatment should target the disease at multiple levels, including reduction of stress pathways, reduction of the toxicity of the diseased white matter, provision of patient derived cured/healthy white matter cells (stem cell transplantation) and possibly gene therapy.
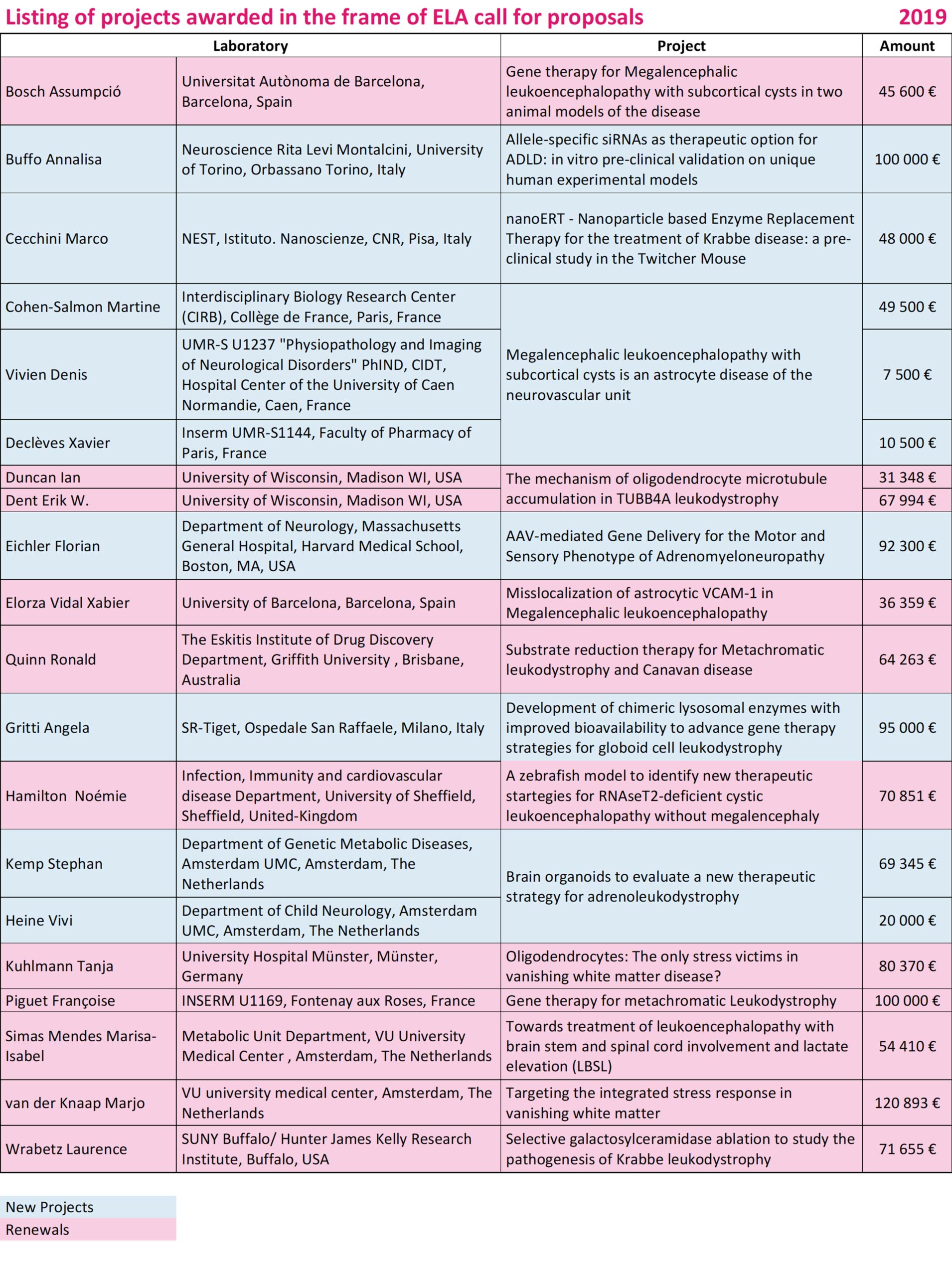
Buffo Annalisa • Italy
2019-006I2
Allele‐specific siRNAs as therapeutic option for ADLD: in vitro preclinical validation on unique human experimental models
The adult onset autosomal dominant leukodystrophy or ADLD, is a genetic, fatal and incurable neurodegenerative disease. It is characterized by a loss of the so-called "white matter" of the central nervous system and is manifested by movement disorders and severe alterations of the autonomic nervous system.
The genetic cause is the presence of three copies, instead of the two normally present, of the gene that contains the instructions to produce the lamin B1 (LMNB1) protein, which belongs to a group of structural proteins (lamins) forming the nuclear membrane of the cell. In patients, Lamin B1 accumulates into the cells causing the neurodegeneration.
Through our project, we will provide the first therapeutic option for ADLD, developing a technique called "allele-specific silencing". By using small molecules of RNA called “siRNAs”, we will be able to "turn off" one of the three copies of the gene, restoring the physiological lamin B1 levels, and in turn avoiding the accumulation of the protein and the disease.
To validate our therapeutic strategy mediated by siRNAs, we will generate two innovative in vitro models based on induced pluripotent stem cells (iPSC) derived from ADLD patients that will allow an authentic "disease-in-a-dish" approach. The iPSCs are a highly versatile tool, since they can be used to recreate in the laboratory different types of cells normally difficult or impossible to obtain from a patient, such as those of the central nervous system. In these models we will test genetic products already developed in our lab to measure efficacy and potency on LMNB1 reduction and the absence of negative or dangerous effects. Our project is intended to pave the way towards a therapy for ADLD and also establish distinctive human ADLD-relevant models and therapeutic approaches that may have great importance for future studies non only on ADLD but also on the physiopathology and therapy of other leukodystrophies and genetic diseases.
Cecchini Marco • Italy
2019‐008I2
nanoERT ‐ Nanoparticle based Enzyme Replacement Therapy for the treatment of Krabbe disease: a pre‐clinical study in the Twitcher Mouse
Krabbe disease (KD, or Globoid cell leukodystrophy) is an autosomal recessive, neurodegenerative disease caused by the deficiency of the lysosomal enzyme galactocerebrosidase (GALC). It is a lethal metabolic disorder, with a frequency of about 1/100000 newborns. The early childhood form represents the 85-90% of cases and the onset of symptoms is between the 3rd and the 6th month after birth. First symptoms are dysphagia, nervousness, hypertonia; convulsions are often present as well. During the advanced state, blindness and deafness show up; then a vegetative state arises which ends with death within the first 1-2 years after birth.
Unfortunately, the systemic administration (e.g. by intravenous injections) of GALC is not effective because of the presence of the blood brain barrier (BBB) that forbids the translocation of bulky proteins like GALC into the central nervous system. No cure is currently available for KD, and treatment is symptomatic and supportive only.
Our strategy to overcome this issue is to exploit active nanoparticles capable of transporting GALC across the BBB. Thanks to a previous pilot study supported of ELA, we demonstrated that with this approach it is possible to achieve GALC activity recovery in the brain of the mouse model of KD, and in cells from KD patients.
With this project, we will perform a complete pre-clinical testing in the KD murine model. An enzyme replacement therapy (ERT) protocol will be optimized based on our nanoparticles to deliver functional GALC via systemic administration into the mouse brain. We will test if this therapy can improve the pathophysiology in terms of: i. life span, ii. prevention/slow-down of neuropathological alterations, and iii. preservation of motor functions.
Given that the materials used in this study are already approved for clinical use, in case of successful project outcome, this research has the potential for a short/medium term clinical translation. Finally, we would like to point out that our methodological approach, here proposed to correct GALC deficiency, is potentially applicable to other lysosomal storage disorders with major brain involvement, such as the Metachromatic Leukodystrophy, by changing the cargo (i.e. the functional enzyme) transported by the nanoparticles.
Cohen-Salmon Martine / Vivien Denis / Declèves Xavier • France
2019‐010C4
Megalencephalic leukoencephalopathy with subcortical cysts is an astrocyte disease of the neurovascular unit
MLC is a rare form of leukodystrophy mainly linked to mutations in the MLC1 gene. Patients with this disease suffer from macrocephaly and motor and cognitive symptoms associated with a progressive myelin degeneration. Why the absence of MLC1 leads to these defects is an open question. MLC1 is a molecule produced by astrocytes, which are the major glial cells of the brain and is enriched at their interface with the blood vessels. Since astrocytes control vascular functions in the brain, we proposed that brain vascular pathological mechanims could be involved in MLC, a question that has never been adressed. Using a mouse model deficient for MLC1, we started to uncover a gliovascular pathology in MLC. Our results suggest that it might be the first pathological sign of MLC. We now propose to further explore these vascular alterations with the underlying idea that knowing their causative mechanisms could provide therapeutic options for MLC patients.
Eichler Florian • USA
2019‐012I2
AAV‐mediated Gene Delivery for the Motor and Sensory Phenotype of Adrenomyeloneuropathy
Adrenomyeloneuropathy is a debilitating lifelong disorder that currently has not treatments available. As an inherited disorder, gene correction is clearly necessary to alter the trajectory of disease burden. We have a developed an approach to deliver a healthy copy of the faulty gene directly into brain and spinal cord.
We have tested this in mice with the disease and have made key insights into a cell type critical to this process: neurons in the brain and alongside the spinal cord. With this knowledge we are now improving gene delivery using new viral vectors.
Importantly we have also developed industry partnerships that can help with manufacturing and setting up future clinical trials. Beyond creating a much-needed treatment, we are also through our studies gaining a better understanding of the disease biology of adrenomyeloneuropathy.
Gritti Angela • Italy
2019‐015I2
Development of chimeric lysosomal enzymes with improved bioavailability to advance gene therapy strategies for globoid cell leukodystrophy
Globoid Cell Leukodystrophy (GLD) is a neurodegenerative lysosomal storage disease (LSD) due to the genetic deficiency of beta-galactosylceramidase (GALC). The rapid disease progression of the infantile forms and the severe neurodegeneration pose major issues for the development of effective treatments. Currently, GLD patients lack real therapeutic options.
The promising but still modest results obtained in pre-clinical models using innovative approaches (i.e. gene/cell therapies) highlight the difficulty of providing timely (before onset of symptoms), pervasive (to all affected tissues), and long-term (ideally for the whole life) therapeutically relevant levels of GALC enzyme in a safe manner. This difficulty relies in part on our imperfect understanding of the mechanisms of enzymatic correction in the different cell types that are targets (i.e brain cells) or effectors (the progeny of blood stem cells) in the context of gene/cell therapy approaches. This is a gap that we aim to fill with this study.
The long-term goal of this study is to design therapeutic approaches based on solid mechanistic ground achieved using relevant GLD models. We hypothesise that the use of a GALC enzyme engineered to increase its secretion and capability to cross the blood-brain barrier may boost the efficacy of gene/cell therapy approaches in GLD, as it does in pre-clinical models of similar diseases. Taking advantage of our expertise in the study and treatment of GLD, and building upon the availability of novel reagents and tools, we will design chimeric GALC enzymes that will be tested for secretion/bioavailablity, safety, and modality of action in direct comparison with the unmodified enzyme in relevant cells types (i.e. hematopoietic stem/progenitor cells and differentiated progeny) and, ultimately, in GLD mice.
Successful completion of this project will increase mechanistic knowledge of disease correction in GLD, paving the way to novel gene/cell therapy strategies using modified enzymes to be applied as independent treatments and/or in combination to achieve global disease correction.
Kemp Stephan • The Netherlands
2019‐020C2
Brain organoids to evaluate a new therapeutic strategy for Adrenoleukodystrophy
X-linked adrenoleukodystrophy (ALD) is the most common leukodystrophy. All ALD patients have a mutation in ABCD1 and accumulate very long-chain fatty acid (VLCFA) in tissues, including brain and spinal cord. In adulthood, virtually all males and >80% of women develop chronically progressive myelopathy (adrenomyeloneuropathy) for which no disease-modifying therapy is available. Hematopoietic stem cell transplantation (HSCT) and ex vivo autologous gene therapy are effective in treating cerebral ALD, but only in the early stages of brain inflammation. Unfortunately, HSCT-treated patients can still develop myelopathy in adulthood, because HSCT is only effective at halting the inflammatory component of the disease without addressing the underlying biochemical defect. This therapeutic gap highlights the need to develop effective treatments aimed at the normalization of VLCFA levels in the brain and spinal cord. Using skin cells from ALD patients we have demonstrated that saturated VLCFA induce cellular stress, with prolonged exposure resulting in cell death. This effect is not observed with mono-unsaturated VLCFA. We identified small-molecules that activate an alternative metabolic route that converts saturated to mono-unsaturated VLCFA.
Treatment of ALD cells completely corrects VLCFA levels. Treatment of the ALD mouse with these molecules added to their food results in a reduction in adrenals, spinal cord and brain. Unfortunately, are these small molecules not specific enough and cause side-effects. We are currently searching for more specific small-molecules. When found, these small-molecules must be tested in an ALD disease model. In this project we will develop a novel ALD disease model.
We already generated stem cells from control and ALD skin cells. These stem cells can be used to generate organoids, which is a miniaturized and simplified version of an organ. Interestingly, with the proper tools we can use these stem cells to generate brain organoids.
The availability of control and ALD brain organoids would be a major step forwards in the development of a therapy for ALD and other leukodystrophies. Organoids will lead to a reduction in animal studies and they are a preclinical model that better reflects the disease.
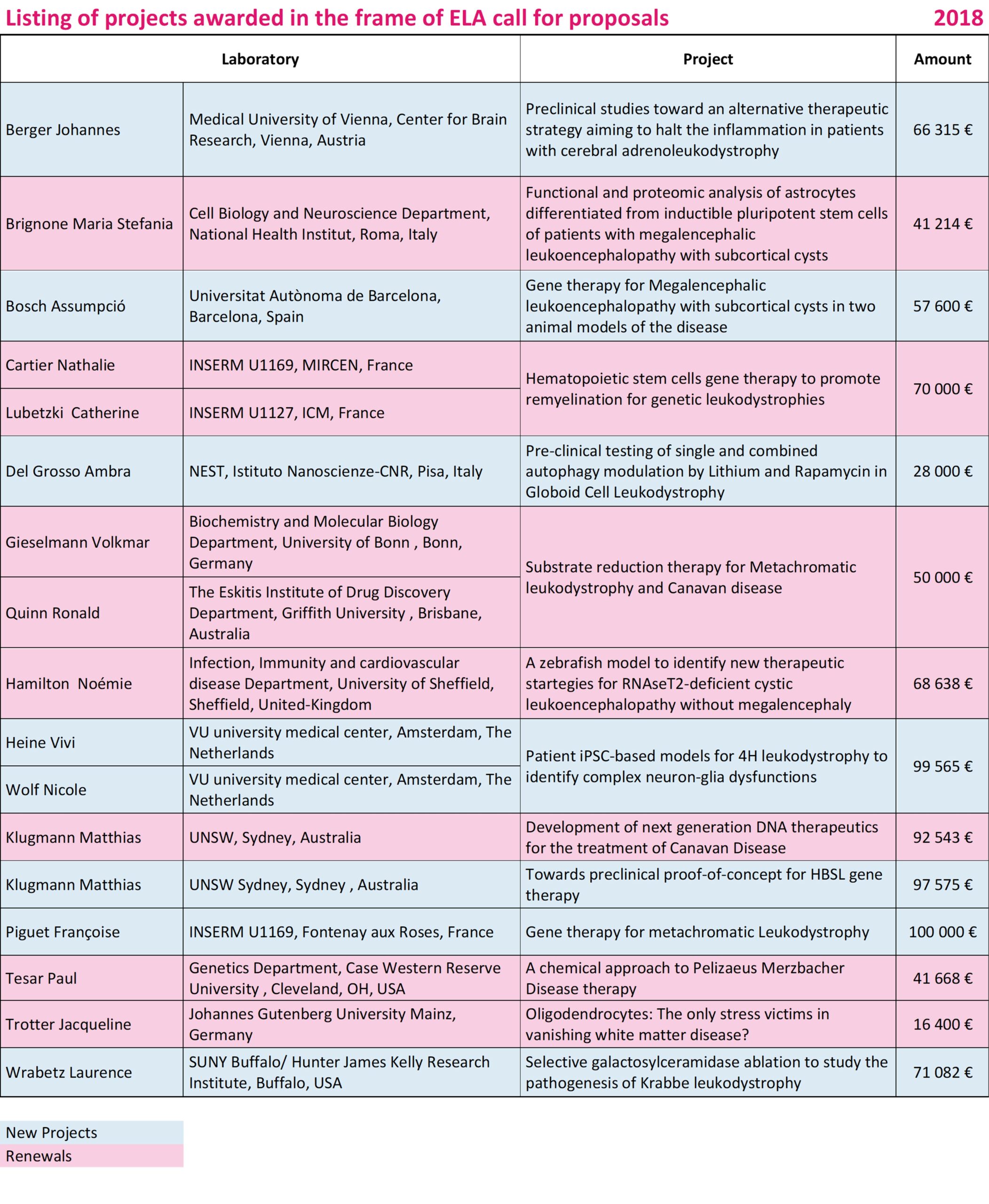
Berger Johannes • Austria
2018-003I2
Preclinical studies toward an alternative therapeutic strategy aiming to halt the inflammation in patients with cerebral adrenoleukodystrophy
X-linked adrenoleukodystrophy (X-ALD) is the most common leukodystrophy with the adult-onset peripheral nerve disease adrenomyeloneuropathy being the underlying core syndrom. However, the majority of patients also develop fatal inflammatory brain demyelination, termed cerebral ALD (CALD). Hematopoietic stem cell transplantation (HSCT) is an established treatment that is able to halt CALD in both childhood and adult patients and is currently, next to hematopoietic stem cell gene therapy (HSCGT), the only available life-saving intervention for CALD patients. Unfortunately, both HSCT and HSCGT are only effective during a narrow therapeutic window at the beginning of CALD, thus excluding patients with advanced CALD from these treatments. We propose to validate pharmacological compounds known as histone deacetylase (HDAC) inhibitors, which are well-tolerated anti-cancer drugs, as a means to stop the macrophage-related inflammatory responses, with a potential ultimate application for advanced CALD patients. We will compare and evaluate the dosage and ability of three different HDAC inhibitors to reverse functional abnormalities like pro-inflammatory activation and accumulation of very long-chain fatty acids in macrophages isolated from the blood of X-ALD patients. X-ALD macrophages are the immune cells most affected by X-ALD and thus, are therapeutic targets. We think that if HDAC inhibitors prove to be effective in X-ALD macrophages, these drugs might delay, ameliorate or even halt the cerebral inflammation in patients with CALD.
Bosch Assumpció • Spain
2018-005I2
Gene therapy for Megalencephalic leukoencephalopathy with subcortical cysts in two animal models of the disease
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a rare genetic disorder characterized by an abnormal head, loss of motor functions, epilepsy and mild mental decline. The disease is caused by mutations in two genes named MLC1 and GLIALCAM. There are no therapies for MLC patients, only palliative treatment. Interestingly, some patients with GLIALCAM mutations show a remitting phenotype, suggesting that it may be possible to improve the phenotype of MLC patients, even at later stages of the disease.
We present here a gene therapy preclinical therapeutic approach for MLC patients using two animal models of the disease, the Mlc1 and the GlialCAM knock-out mice. Therapeutic genes will be delivered by one-time treatment to the CNS, overcoming the difficulty of crossing the blood-brain barrier. We hope that the results of this project will be able to provide the first therapeutic tools for patients affected with MLC and may have also implications to treat other diseases affecting the myelin.
Del Grosso Ambra (PI: Marco Cecchini) • Italy
2018-008F2 (Fellow)
Pre-clinical testing of single and combined autophagy modulation by Lithium and Rapamycin in Globoid Cell Leukodystrophy
Globoid Cell Leukodystrophy (GLD) is a rare, hereditary disorder (with a frequency of about 1/100000 newborns) triggered by a deficit of the lysosomal enzyme galactosylceramidase (GALC) and characterized by the accumulation of galactosylsphingosine (psychosine, PSY) in the nervous system. PSY is a cytotoxic sphingolipid, which leads to the widespread degeneration of oligodendrocytes and Schwann cells, causing demyelination. Little is known about the molecular mechanisms by which PSY imparts toxicity and there is currently no cure available for GLD. The early-infantile and most widespread form of this lysosomal storage disorder (LSD) is degenerative, rapidly progressive and lethal. Bone marrow transplantation is currently the only clinically applied method to treat GLD, but gene therapy has yielded good results in experimental models. However, the recent literature strongly suggests that GALC-deficiency correction is not sufficient to completely rescue the GLD phenotype. Thus, supportive therapies
specifically addressing secondary targets of the disease might be needed to improve the final therapeutic outcome.
Autophagy, although much studied for many LSDs and neurodegenerative diseases (NDs), has been poorly investigated in GLD. Autophagy dysregulation has been only recently demonstrated, by us and others, in two in vitro GLD cell models.
Here, given our previous results and the wide data form literature showing autophagy activation as a promising therapeutic strategy for NDs and LSDs, we propose the pre-clinical testing of two autophagy activators in the naturally occurring murine model of GLD, the Twitcher mouse (TWI). Specifically, we aim to test in the TWI mouse the effects of Lithium, Rapamycin and of their combination, studying the rescue of a complete set of behavioural and biochemical parameters.
Certainly, this study will be very useful to deeply understand the role of autophagy in the molecular pathogenesis of GLD. Furthermore, in case of positive results, therapy will be readily applicable to humans for clinical testing, thanks to the fact that both tested drugs are already available as pharmaceutical preparations. Therefore, in a forward-looking vision, Lithium and/or Rapamycin could be used in combination with a main GALC-deficiency correcting therapy (as gene therapy and/or enzyme replacement therapy) to help in the achievement of a complete GLD phenotype rescue.
Heine Vivi / Wolf Nicole • The Netherlands
2018-011C3
Patient iPSC-based models for 4H leukodystrophy to identify complex neuron-glia dysfunctions
4H syndrome, an inherited brain white matter disorder (leukodystrophy), leads to considerable clinical handicap, ranging from mild to severe. Most patients deteriorate over time. There is no treatment yet, and we do not understand what goes wrong in the brain. There are no good animal models for this disease, hampering research. We think that besides white matter and myelin, also the nerve cells (neurons, also called grey matter) and their processes (axons) are damaged from early disease stages on. In our project, we want to explore this grey matter involvement with an innovative approach.
We are able to make white and grey matter cells from 4H patient and control fibroblasts. Earlier studies showed that 4H patient cells show affected expression of genes involved in nerve cell development. As nerve cells provide signals to white matter cells and myelination, neuronal defects could underlie abnormalities found in grey and white matter areas in 4H patient brains. We want to study co-cultures of both grey and white matter cells derived from patient cells to know how they influence each other. In the future, this might be a good model for drug screening to treat 4H.
Klugmann Matthias • Australia
2018-014I2
Towards preclinical proof-of-concept for HBSL gene therapy
Hypomyelination with Brain stem and Spinal cord involvement and Leg spasticity (HBSL) is a leukodystrophy caused by defective cytoplasmic Aspartyl-tRNA synthetase (DARS). This enzyme is involved in building proteins, a fundamental biological process in bacteria and man alike. HBSL is caused by autosomal recessive mutations in the DARS gene and all point mutations identified result in neurological disease. HBSL is a potentially fatal spectrum disorder with no treatment and unclear etiology. In a pioneering effort our team at the University of New South Wales has generated the first mouse model of HBSL by introducing the same mutations causing HBSL in patients into the mouse Dars gene. Accurate animal models are the prerequisite to study the disease mechanisms and to develop and test treatments. Moreover, we have already identified that DARS expression in the brain of both mouse and man is enriched in neurons with far less expression in glia. This suggests that neurons might be the cells that a first line HBSL therapy should ideally target. In the first aim of this research proposal, we plan to engineer more mouse models that genetically mimic HBSL mutations and characterize them in order to model different forms of severity of HBSL. The second aim will be to develop a gene therapy platform for expression of a healthy copy of the DARS gene in mice. These experiments will help to identify the optimal route of delivery and the best timing for intervention. In the third aim we will perform a proof-of-concept gene therapy in the most relevant mouse model using optimized paramemters for DARS gene therapy identified in the previous aims. This project will yield an accurate animal model of HBSL that will be instrumental for preclinical testing of gene therapy or other treatment avenues. We propose that this study will generate data with high clinical relevance as our preclinical endpoints and gene therapy platform could easily be adapted for the treatment of other leukodystrophies caused by abnormal protein translation.
Piguet Françoise • France
2018-019I2
Preclinical studies specifically testing therapies (gene, cell, enzyme or pharmacologic therapies) for leukodystrophies
Metachromatic Leukodystrophy (MLD) is a rare demyelinating disease, due to arylsulfatase A (ARSA) deficiency, an enzyme involved in the catabolism of sulfatides, the main component of the myelin sheath. This deficiency leads to progressive demyelination of central nervous system and peripheral neuropathy. The most frequent form of the disease is the late infantile form of the disease which is characterized by a rapid progression of the disease, especially after the first onset of symptom. Ex vivo gene therapy develop by the group of Alessandra Biffi, and based on engraftment of lentiviral transduced hematopoietic stem cells has been shown to be effective for presymptomatic forms of MLD but not effective in early symptomatic patients, probably due to the rapid evolution and the time needed for the engrafment. On the other hand, enzyme replacement was shown as potentially effective but needed a chronic delivery. In the group, we previously proposed a gene therapy approach for MLD based on intracerebral delivery of an AAVrh.10 encoding ARSA. We had established proof of concept in mouse model of the disease and scale up study in non-human primates (NHP) leading us to propose a clinical trial which included 4 patients from 2013 to 2016. Despite ARSA expression in the brain and detection in cerebrospinal fluid (CSF) we failed to obtained any therapeutical benefits in these patients. Our point is that expressing ARSA in the CNS is fundamental to rapidly stop the disease progression, however, it could be essential also to bring back in patient healthy microglial cells. This project aim at establishing the proof of concept of rapid, sustained and important ARSA expression in the whole CNS (brain, spinal cord) and potentially also in the peripheral nerve. To achieve this aim, we propose to use a novel serotype of AAV: AAVPHP.eB that has been engineered to efficiently cross the blood brain barrier (BBB) after IV delivery. The project will be divided in two main parts, first a rapid study in the MLD mouse model both for pre and post symptomatic treatment, in order to evaluate efficiency of AAVPHP.eB-ARSA vector for biodistribution, ARSA expression, sulfatides correction, Purkinje cell prevention and behavioral testing. In a second phase, we want to test this vector in NHP thought several routes of delivery: intravenous (IV), intrathecal (IT) and intracerebroventricular (ICV), especially by potential coupling of these routes of administration to optimize CNS targeting. This with the final aim to propose a clinical trial for symptomatic MLD patients.
Wrabetz Lawrence • USA
2018-023I4
Selective galactosylceramidase ablation to study the pathogenesis of Krabbe leukodystrophy
Krabbe leukodystrophy (KD) is a progressive and fatal neurologic lysosomal storage disorder that usually affects infants and causes death before two to three years of age. Hematopoietic Stem Cell Therapy extends the long-term survival with improved quality of life for KD patients, but it is not a cure. Using our newly developed KD animal model, we will identify which brain cell(s) needs to be efficiently cured with therapy.
